
Ivermectin's Universe
March 16 2022


As a follow-up to “Ivermectin’s Little Secret”, it should be appreciated that while the protein-protein ivermectin interaction pathway described by Li N et al (2021) for SARS-CoV-2 proteins gives us a glimpse of what ivermectin can do in terms of direct interaction with SARS-CoV-2 (Figure 1), it is important to understand that ivermectin has a broader effects on cells; for example where the core pathways involve EGFR, EPO, CXCL8 and IL6 (Figure 2).
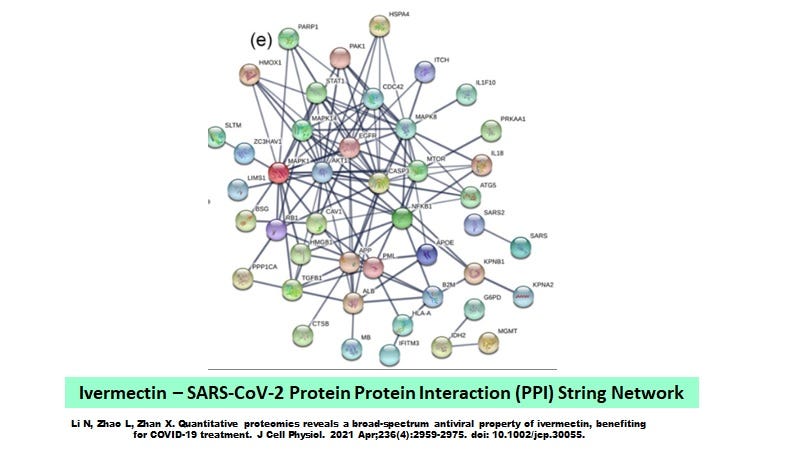
Figure 1. The Ivermectin - SARS-CoV-2 PPI Network

Figure 2. The Ivermectin Network Involves CORE Molecules EGFR, IL6 CXCL8 and EPO
EPO (erythropoietin) stimulates the bone marrow to produce more red cells to carry oxygen which is quite relevant to COVID-19 severity where oxygenation of the blood is poor. There is also talk of ivermectin as an anti-malarial drug putatively due to ‘endectocide’ activity causing the death of the Anopheles mosquitoes (Slater HC et al., Lancet Infectious Diseases, 2020; Alonso P & Engels D. Malar J, 2017). Of course EPO features in the hypoxia (HIF-1) pathway and which interestingly stimulates foam cell formation, which involves EGFR.
CXCL8 (also known as IL8) attracts neutrophils to the site of infection and is a potent angiogenic factor. Like IL6 it is also involved in sepsis. In its pathway, it connects to CCL2 (attractant for monocytes) and to ERBB2 (part of the EGFR family and which may cause epithelial mesenchymal transition (EMT), the malignant phenotype ) and to CREBBP as part of the stress response. CCL2 also connects to IRF-1 which is the main driver of the LPS/TLR4 response which strongly induces the most inflammatory molecule around, ACOD1 (Immunoresponsive Gene 1; IRG-1). IRF-1 activity may be blocked by VDR (Tallam A et al., PLoS One, 2016).
IL6 is a potent inducer of the acute phase response and induces ZBTB16 needed for myeloid maturation and differentiation. The overexpression of IL6 is associated with COVID-19 severity. In the glucocorticoid pathway network, AFP interacts with IL6 which interacts with CXCL8.
COVID-19 Autopsy Tissue Findings in the Lungs
If you recall, we previously talked about how in severe patients with COVID-19, only the macrophages in bronchoalveolar lavage fluids (BALF) contained SARS-CoV-2 RNA and this RNA was replicating (Ren X et al., Cell, 2021). In contrast, cells in PBMC did not have SARS-CoV-2 transcripts nor did BALF samples from moderate COVID-19 patients. These infected myeloid cells did not express ACE2 nor TMPRSS2 but did overexpress BSG (inducer of foam cell formation) and the transferrin receptor (TFRC) both implicated as co-factors for SARS-CoV-2 entry. While Ren et al suggested a role in a subclass of B cell plasma cells in mediating severity of COVID-19 (the B_c05-MZB1-XBP1 which had sequences which overlapped with known SARS-CoV-2 neutralizing antibodies, suggestive of ADE), FCGR2A (implicated in ADE) was not found on the infected macrophages. On the other hand for total cells infected with SARS-CoV-2, FCGR2A was upregulated on virus infected cells in PBMCs and in BALF.
Ren et al, did not address the discrepancy that infected macrophages did not seem to overexpress FCGR2A nor did they clarify what cells were infected by SARS-CoV-2 identified in both BALF and PBMCs which expressed FCGR2A.
However, when Delorey TM et al (Nature July 2021) examined autopsied lung tissues we seem to have a more complete picture (Figure 3). Like the sBALF findings by Ren et al, myeloid cells contained most of the SARS-CoV-2 transcripts yet did not express ACE2 nor TMPRSS2. As shown in Figure 4, the highly inflammatory CD14+CD16+ monocytes (CCR5 positive, see Table 1 attached) % showed the highest level of number of cells infected (about 40 cells positive).
This is exactly the same cell type that founder/transmitter ‘R5’ strains of HIV-1 (another enveloped pandemic RNA virus) need to infect in order to colonize the host (Crowe S et al., J Leukocy Biol, 2003; Shen R et al, J Virol, 2009). Importantly, this implies pandemic RNA viruses may need to commonly overcome trained innate immunity mediated by CD14+CD16+ CCR5+, monocytes which also co-express Fc receptors CD32 (FCGR2A), CD16 (FCGR3A) and/or CD64 (FCGR1A) which are the cell types capable of producing HERV-K102 protector particles.
Most remarkably, all myeloid cell types were infected.
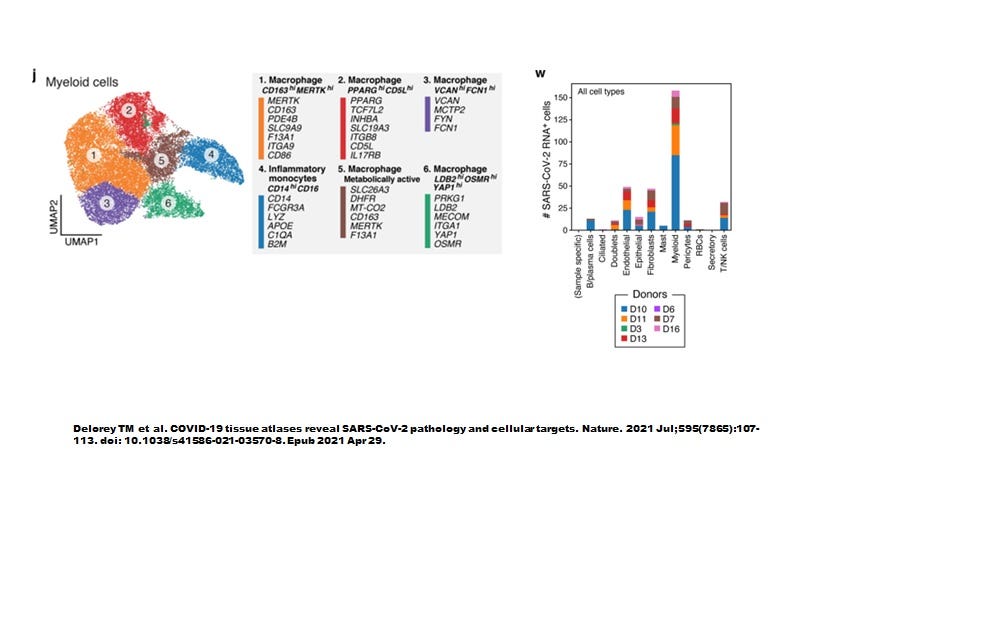
Figure 3. Six types of myeloid cells in autopsy lung materials. (Note that neutrophils were absent and the authors suggested this had somehow to do with death of the host).
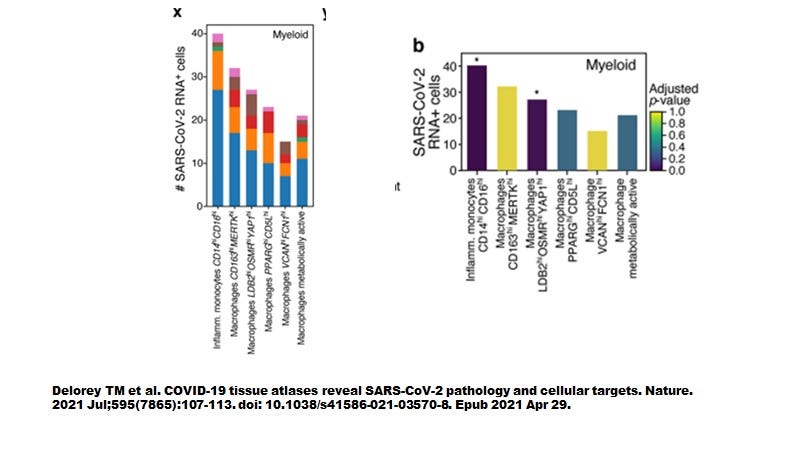
Figure 4. All myeloid cells from autopsy lung tissues are infected by SARS-CoV-2 and around 40% of inflammatory monocytes were infected.
Delorey et al only provided details on the differentially expressed genes (DEGS) for the highly inflammatory monocytes and the PPARG (hi) CD5L (hi) macrophages (see summary of selected DEGs in attached Table 1). When the pathways were superimposed on the ivermectin-SARS-CoV-2 PPI network (Figure 5), we see that the three MAPKs are stimulated in the macrophages while only MAPK14 (p38) was stimulated in the monocytes.
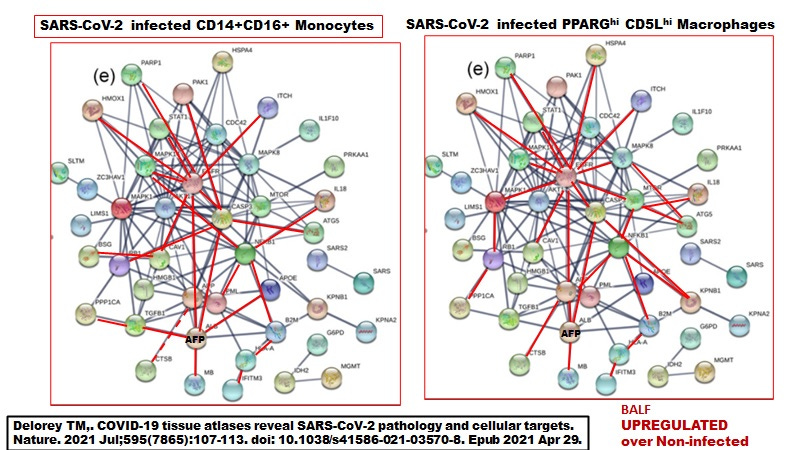
Figure 5. Macrophages but not monocytes in autopsy COVID-19 lung tissues involve activated MAPK8 and MAPK1.
Recall that MAPK8 may be upregulated associated with conversion of LB (negative) M1-like foamy macrophages to M2 LB positive foamy macrophages blocking HERV-K102 particle production. On the other hand, MAPK1 may block foam cell production (Figure 6). Apparently, vitamin D deficiency promotes the conversion to the M2 (anti-inflammatory macrophages) and it was suggested that ivermectin along with vitamin D, may block this transition.
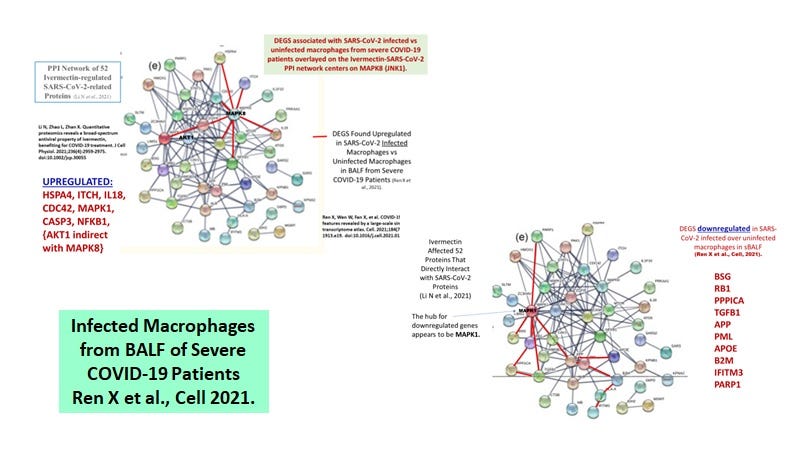
Figure 6. MAPK8 may be upregulated associated with conversion of LB (negative) M1-like foamy macrophages to M2 LB positive foamy macrophages blocking HERV-K102 particle production. MAPK1 may additionally block foam cell production. Data from BALF of severe COVID-19 Patients (Ren X et al., Cell 2021).
Putatively, HERV-K102 Particles may be Upregulated With COVID-19 Severity
As was suggested in a previous post, extracellular vesicles correlate with COVID-19 severity (about 10 (9) per ml of serum) (Maugeri N et al., 2021) and it was implied these might be HERV-K102 particles (Figure 7).
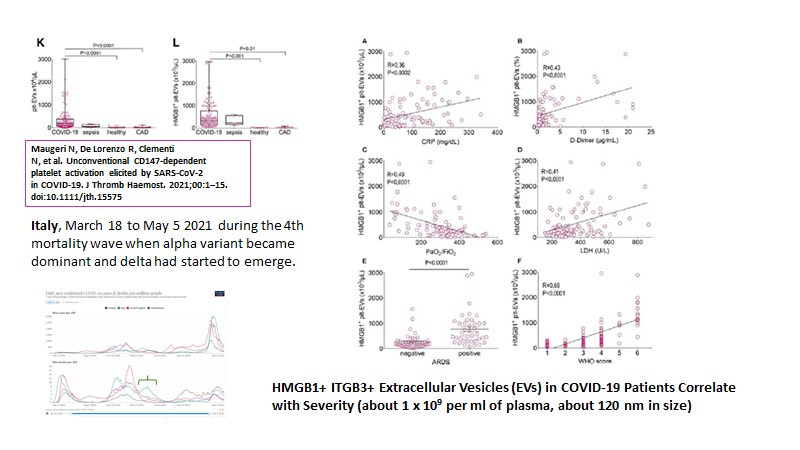
Figure 7. Extracellular Vesicles (suggestive of HERV-K102 particles) correlate with various indicators of COVID-19 Severity.
Which Cells are Producing and Releasing HERV-K102 Particles by Lysis?
By examining the ivermectin - SARS-CoV-2 PPI pathways in Figures 5 & 6, it would appear that only the inflammatory monocytes and not the macrophages would be capable of producing and thus releasing HERV-K102 particles. The activation of MAPK8 and MAPK1 only in the macrophages would favor M2 FM over M1 (MAPK8) and downregulate particle induction by downregulating BSG, (MAPK1 and see Table 1 attached). So while both cell types are infected with replicating SARS-CoV-2 (see Table 1 attached), it is likely only the inflammatory monocytes that produce HERV-K102 particles.
It is well established that HERV-K102 envelope becomes expressed on virally infected cells or tumor cells but not normal cells and that patients with tumors or viral infections often have innate antibodies reactive to HERV-K102 envelope (reviewed in Laderoute M. Clues to finding correlates of risk/protection for HIV-1 vaccines. F1000 Research 2018, 6:868. https://doi.org/10.12688/f1000research.11818.2). Antibodies to HERV-K102 envelope are found with virus infections but especially as concerns a pandemic RNA virus, HIV-1 (Laderoute M et al., AIDS, 2007). Around 80% of HIV-1 positive patients have high levels of HERV-K102 envelope antibodies. These innate ‘autoantibodies’ can directly activate the cell death machinery (CIDEA and/or CASP8 to CASP3) to mediate the killing of HERV-K102 envelope expressing cells (Wang-Johanning F et al, JNCI 2012). Notably, both the SARS-CoV-2 infected monocytes and macrophages highly express these 3 death induction genes (see Table 1 attached). However, apoptosis resistance may be mediated in part by ERBB2, which is down modulated in the monocytes but upregulated in the macrophages. This means the monocytes are more capable of undergoing apoptosis than the macrophages. Moreover, MX1 is a marker of macrophages/sebocytes committed to the induction of apoptosis and again it is only upregulated in the SARS-CoV-2 infected monocytes but not the macrophages (see Table 1 attached). Thus, it is likely that the highly inflammatory monocytes are the cells in autopsy lung tissues which are producing and releasing HERV-K102 protective particles. The question remains, do these antibodies to HERV-K102 envelope help the release of HERV-K102 particles from the SARS-CoV-2 infected inflammatory monocytes?
As mentioned in the post of March 9, 2022, Dispinseri S et al (Nature Commun, May 2021) described innate neutralizing antibodies that developed during the first 2 to 4 weeks after symptom onset that correlated with a reduced risk of death and faster clearance of SARS-CoV-2 from the nasopharynx. In contrast the SARS-COV-2 specific adaptive immunity antibodies did not develop until week 5. It should be noted that COVID-19 patients who did not develop these innate neutralizing antibodies were at high risk of death.
So it is possible that for non-severe patients the antibodies to HERV-K102 help to clear SARS-CoV-2 from sebocytes in the nasopharynx as well as block infection by neutralizing SARS-CoV-2 particles. Recall that SARS-CoV-2 like HIV-1 is an enveloped RNA virus so when it buds from the cell surface it will pick up the HERV-K102 envelope expressed on the surface of virally infected cells. This then allows the antibody to ‘neutralize’ the SARS-CoV-2 particles. Similarly for infected monocytes/macrophages, the antibodies may shorten the time to lytic release. However, it remains unknown if severe patients may be at additional risks due to lower HERV-K102 antibody levels. It would be a simple matter to test for these antibodies at different stages of COVID-19 severity. It just has not been done, yet.
Scenario for Innate versus Adaptive Immune Responses in COVID-19
Putting the pieces of the puzzle together, it would seem those without co-morbid conditions and not experiencing pre-existing immunosenescence, would rapidly release HERV-K102 particles systemically and develop the protector HERV-K102 envelope antibodies by day 10 post symptom onset (see previous post). This would clear the viral infection and reduce the risks of severe COVID-19 disease by avoiding seroconversion that occurs around day 12 (Chvatal-Medina M et al., Frontiers in Immunology, April 2021). However, in those patients unable to achieve this, they may instead develop adaptive antibodies to SARS-CoV-2 (ie., seroconvert) including neutralizing antibodies against spike protein. With time there would be selection of immune escape mutants in the host by the spike specific antibodies. This then brings on the risk of antibody dependent enhancement (ADE) mediating monocyte/macrophage infection, which according to Ren et al (2021) only happens in severe COVID-19 patients. As we see in Figures 5 and 6, SARS-CoV-2 infection putatively knocks out the HERV-K102 protector system in macrophages while at the same time the newly recruited monocytes undergo a magnified inflammatory response (featuring ACOD1/IRG-1 associated with elevated IL6, TNF-alpha, IFN-gamma, CXCL8, CRP, TLR4 activation and release of S100A8, IDO1, C1QA and made worse by the downregulation of VDR due to insufficient vitamin D, Table 1). Both Ren et al and Delorey et al report that it is the inflammatory newly recruited CCR5 positive, CD14+ CD16+ monocytes in the lungs that are responsible for cytokine storm. These infected monocytes probably release HERV-K102 particles by lysis. (The latter would tend to provide feedback inhibition of the inflammatory response if the virus was cleared). At the point where 40% of the inflammatory monocytes are infected by SARS-CoV-2, the patient dies, despite the presence of high levels of presumptive HERV-K102 EV particles (Maugeri N et al, 2021).
Along these lines, unlike the findings by Ren X et al, 2021, Delorey TM et al (Nature, 2021) are reporting high levels of FCGR2A and FCGR3A in both the infected monocytes and macrophages in the post-mortem lungs of deceased COVID-19 patients (Table 1 attached). These are the receptors that mediate ADE. Thus, when taken altogether, this appears to suggest that ADE mediated by adaptive immunity antibodies to spike protein may be responsible for the death of COVID-19 patients due to the widespread infection of myeloid cells.
It should be noted that this contrasts sharply with the contention of Dispinseri et al who erroneously claimed the adaptive immunity antibodies to SARS-CoV-2 spike protein “protected against death” when in fact their evidence showed innate antibodies mediated protection. Researchers invariably report that spike protein antibodies and/or NAbs are associated with COVID-19 severity and not protection, and is more common with advancing age, male sex and comorbidities (reviewed in Chvatal-Medina M et al, Frontiers in Immunology, April 15, 2021).
Summary and Conclusions
In summary, we now have the evidence to support the contention that 1) ADE could be associated with the loss of the critical HERV-K102 host defense in macrophages and 2) SARS-CoV-2 infection may induce the induction of a high state of inflammation in the CCR5 positive CD14+CD16+ newly recruited monocytes. In either case this data implies ADE may be directly responsible for infection of myeloid cells and relates to COVID-19 deaths. Notably the infection rate of up to 40% of the inflammatory monocytes in the lungs being infected by SARS-CoV-2 is associated with COVID-19 patient demise.
Sweeney TE et al, (Crit Care Med, 2021) have described DEGS which can be used to categorize sepsis patients into the following groupings as validated for COVID-19 patients:
Inflammopathic: ARG1, LCN2, LTF, OLFM4, and HLA-DMB with a death rate of about 18%;
Adaptive: YKT6, PDE4B, TWISTNB, BTN2A2, ZBTB33, PSMB9, CAMK4, TMEM19, SLC12A7, TP53BP1, PLEKHO1, SLC25A22, FRS2, GADD45A, CD24, S100A12, and STX1A with a zero % death rate; and
Coagulopathic: KCNMB4, CRISP2, HTRA1, PPL, RHBDF2, ZCCHC4, YKT6, DDX6, SENP5, RAPGEF1, DTX2, and RELB with a death rate of about 43%, the highest of all the groups. All of these DEGS are upregulated in blood except for DDX6 which is strongly downregulated with the coagulopathic category. Interestingly DDX6 is needed for the transport of HERV-K102 genomic copies to the cytoplasm for incorporation into particles (Manghera M et al., J Virol, 2016) and thus it is not surprising that this category has the highest mortality rate (reflecting the highest loss of protective innate immunity by HERV-K102 particles).
Ren et al, have suggested the dominating macrophage population in the BALF of severe COVID-19 patients falls into the MO_c1 category with higher TGFB1, PPARG and NR1H3 where the last two are considered anti-inflammatory and where TGFB1 is considered profibrotic as demonstrated with ARDS and ALI for SARS infections (Gralenski LE et al, mBio 2013). This may be why Delorey et al only provided details on the differentially expressed genes (DEGS) for the highly inflammatory monocytes and the PPARG (hi) CD5L (hi) macrophages (see summary of selected DEGs in attached Table 1). Both cell types in the autopsied lungs from COVID-19 patients however have DDX6 downregulated (Table 1). This raises the possibility that these COVID-19 patients may have died due to hypercoagulopathic symptoms.
In closing, while omicron was thought to be a less pathogenic variant than delta as witnessed in the slow to vaccinate nations, in the fast vaccination nations, the omicron COVID-19 deaths per million (standardized against the world average) were notably increased over delta (see previous blog). This means that vaccination with two dose (or more) mRNA vaccines which stimulates SARS-CoV-2 spike specific antibodies and especially neutralizing antibodies may be contributing to the death toll despite protection by adaptive T cell responses by the vaccines. In contrast, nations that allowed natural infections with the delta variant, were better protected against omicron deaths.
These findings bring to light the potential deadly nature of spike specific antibodies especially the neutralizing antibodies in their potential role in mediating ADE. The time is now to STOP using adaptive immunity vaccines which generate spike specific antibodies that may jeopardize the host’s innate protection system through ADE and which instead mediate cytokine storm and an increased risk of death. One also needs to consider that omicron and delta specific vaccines may need to be banned even from clinical trials. Clearly mandating adaptive immunity vaccines which are contributing to the rising death toll with omicron and later variants, should no longer be tolerated.
Thank you for writing on substack. I wish I could understand the details of all of what you have written :)! I would second the request for you to weigh in on the micro clotting issue. We are seeing a lot of ppl post C19 with dyspnea; failing six minute walks etc. even after decent treatment regimes. Some are gradually getting better, others struggle still. As a nurse, it feels like I am in a slow motion film and the population is slowly being disabled before my eyes. Looking for pathways of healing for these folks.
The basis of SARS-2 mortality is blocking interferon-1 which unregulated HERV-K102, in the absence of which causes ADE from infected monocytes and neutrophils. Wow! Isn’t nature ironical to use viruses to kill viral infections. Perfect!